
5 Signs Your Quality System Is Holding Your Organization Back
Your quality system exists to protect your organization. It's supposed to be the infrastructure that keeps products safe, processes consistent, and regulators satisfied. But for a lot of quality leaders in regulated healthcare and life sciences, the system itself has quietly become the problem.
Not because anyone made a bad decision. Most quality systems were built the right way for the size and complexity of the organization at the time they were implemented. Spreadsheets worked when you had twelve staff and one site. A shared drive made sense before your document library grew to three hundred controlled SOPs. Manual training logs were manageable before you had a new product line, a new regulatory requirement, and two audits scheduled in the same quarter.
The system didn't fail. It just didn't grow with you. And at some point — often gradually, sometimes suddenly during an inspection — the gap between what your quality system can handle and what your organization actually needs becomes impossible to ignore.
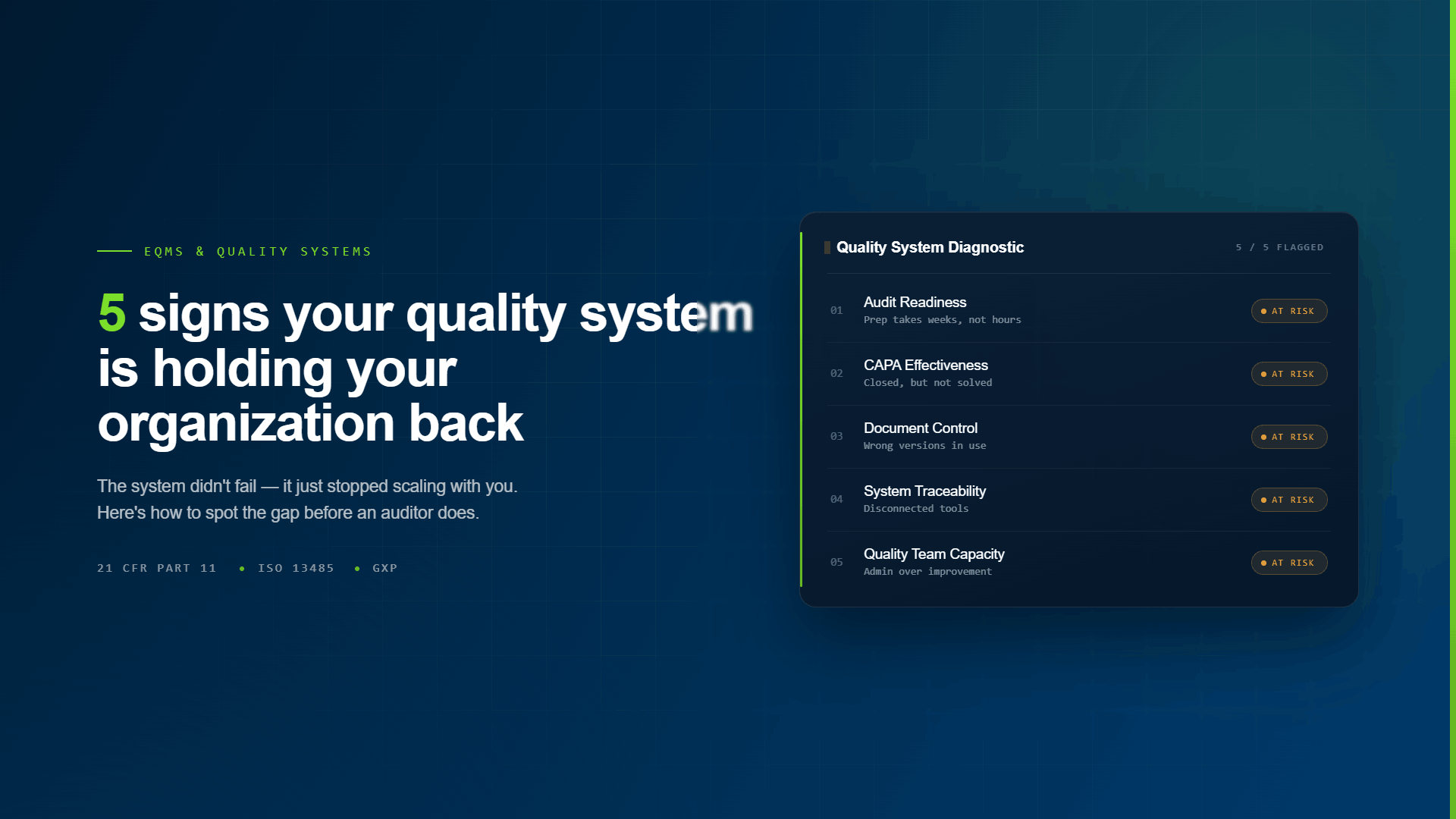
Here are five signs that gap has already opened.
Sign 1: Audit Prep Takes Weeks, Not Hours
Walk into any quality department the month before a scheduled FDA inspection, AABB assessment, or ISO 13485 surveillance audit, and you'll usually find the same thing: staff pulling folders, cross-referencing training records against SOP versions, manually verifying that every CAPA from the last cycle has a closed effectiveness check, and assembling evidence packages that should already exist in a format auditors can review.
If your audit prep looks like this, your quality system is not doing its job.
A functional quality management system maintains a continuous, inspection-ready state. Every document has a version-controlled audit trail. Every training acknowledgment is linked to the document version in effect at the time. Every CAPA has a timestamped record of initiation, investigation, corrective action, and effectiveness verification — all accessible in a single search. When an auditor asks for evidence, you retrieve it in seconds, not days.
The pre-audit fire drill is one of the most normalized dysfunctions in regulated organizations. It's so common that most teams treat it as inevitable. It isn't. It's a symptom of a quality system that doesn't maintain audit readiness automatically — and a sign that your current infrastructure is creating risk instead of reducing it.
The real cost: Beyond the staff hours consumed, extended audit prep is a sign that your records have gaps your team is scrambling to paper over. That's the scenario that produces FDA 483 observations and warning letters.
Sign 2: CAPAs Are Getting Closed Without Actually Being Solved
CAPA is the core corrective engine of any quality system. When it's working, CAPAs drive real process improvement — root cause is identified, corrective and preventive actions are implemented, and effectiveness verification confirms the problem doesn't recur. When it isn't working, CAPAs become a documentation exercise.
Signs that your CAPA process has drifted into the latter:
Repeat findings. If the same deviation, complaint category, or audit observation keeps generating new CAPAs, the root cause was never actually addressed. It was classified, documented, and closed — but not solved.
Missing effectiveness verification. CAPAs that are closed without a completed effectiveness check are one of the most common FDA 483 observations in regulated organizations. It's not a paperwork issue; it's a sign that the system isn't enforcing the process.
CAPA backlog. A growing queue of open CAPAs is a signal that either the root cause investigation step is becoming a bottleneck, or that the corrective actions being assigned aren't actionable enough to get closed. Either way, open CAPAs represent unresolved risk sitting in your quality system.
Disconnected from the rest of the system. If your CAPAs don't automatically link to the deviations, complaints, or audit observations that generated them — and if they don't automatically trigger document revisions or retraining when actions require it — your CAPA management isn't functioning as a system. It's functioning as a task list.
A CAPA management process that produces closed records without verified outcomes isn't protecting your organization. It's generating documentation that looks compliant while the underlying process risk remains unaddressed.
Sign 3: Staff Are Working from Wrong Document Versions
This is one of the most dangerous quality system failures in regulated environments, and it's almost always invisible until an audit or an adverse event surfaces it.
It happens because document control in most organizations relies on people to access the right version — not on the system to enforce it. An SOP gets revised and approved, but someone on the floor is still working from a printed copy of the previous version because the notification didn't reach them, the training wasn't completed, or the old document wasn't physically removed.
In regulated healthcare and life sciences, this isn't a procedural nuisance. It's a patient safety issue and a regulatory violation. FDA 21 CFR Part 820, ISO 13485, and GxP requirements are explicit: obsolete documents must be prevented from unintended use. An organization that can't demonstrate that control is one that will struggle to explain the deviation that results from it.
The root problem is that many document control systems are built on shared drives, SharePoint sites, or legacy platforms that provide version storage but not version enforcement. The document exists in the right place. But the system doesn't prevent staff from accessing the old version, doesn't automatically identify who needs retraining when a document changes, and doesn't generate the linked training assignment when a new version is approved.
If your document control process depends on people remembering to use the current version, your document control isn't controlled. The system needs to enforce it — automatically, at the version level, with audit trail documentation that proves the right version was in use at the time of any quality event.
Sign 4: You Have Multiple Systems That Don't Talk to Each Other
This is the architecture problem that grows silently behind almost every quality management dysfunction in mid-sized regulated organizations. It usually looks something like this:
Documents live in SharePoint. Training records are in an LMS. CAPAs are tracked in a spreadsheet. Deviations are logged in a different system — or in email. Equipment calibration records are in a standalone database. Audit management is in a shared folder with PDF checklists.
Each system was added to solve a specific problem. And individually, each one probably does what it was implemented to do. The failure isn't in any individual tool. The failure is in the gaps between them.
When your document control system doesn't know that a CAPA required a process change, the document revision doesn't get initiated. When your CAPA system doesn't know which deviation triggered the investigation, root cause analysis starts from a narrower data set. When your training system doesn't know which document version was in effect when a deviation occurred, you can't reconstruct the compliance picture an auditor needs. When your equipment calibration records don't link to the QC data generated by that equipment, your out-of-specification investigation starts with a manual traceability exercise instead of an automated link.
The FDA's expectation — and ISO 13485's expectation — is that your quality system functions as a system. Not as a collection of tools. Traceability requirements assume that records are connected. CAPA effectiveness assumes that corrective actions can be verified against the processes and documents they were supposed to change.
A quality management ecosystem held together by manual hand-offs between disconnected systems will consistently produce the audit finding organizations fear most: incomplete records, gaps in traceability, and evidence packages that can't be reconstructed efficiently under inspection pressure.
Sign 5: Your Quality Team Spends More Time on Administrative Work Than Improvement Work
This is the sign that quality leaders usually feel most acutely but find hardest to quantify for leadership. The work that your quality team was hired to do — identifying process risk, driving systemic improvement, building a quality culture, analyzing trend data across deviation categories — keeps getting crowded out by the work of managing the system itself.
Chasing training completions. Following up on overdue CAPAs. Manually updating version logs. Generating compliance reports by pulling data from multiple systems and consolidating it in a spreadsheet. Rebuilding the same audit evidence package every inspection cycle from the same disorganized source material.
This administrative overhead isn't just an efficiency problem. It's a quality risk.
When your quality team doesn't have time to review deviation trends, the systemic issues don't get surfaced until they become 483 observations. When CAPA owners are chased for updates instead of the system enforcing deadlines, CAPAs slip. When training compliance data requires manual aggregation, the gaps don't get caught until an auditor finds them.
The purpose of a quality system is to create the infrastructure that lets your quality team focus on quality — not on managing the system. When the system itself is the biggest consumer of quality team time, the organization's entire quality function is operating at reduced capacity.
What a Quality System Should Actually Do
A modern eQMS doesn't just store records. It enforces processes, maintains traceability, surfaces risk, and keeps the organization in a continuous state of inspection readiness without requiring manual overhead to sustain it.
That means document control that prevents obsolete versions from being used — not just stores the correct version in a folder. CAPA management that enforces every step from root cause to effectiveness verification — not just tracks the record. Training management that automatically identifies who needs retraining when a document changes — not just logs completed assignments. Deviation management that links every quality event to every related record across the system — not just logs the incident.
And it means those modules working together. A single linked data model where a deviation generates a CAPA, the CAPA triggers a document revision, the document revision generates training assignments, and every step is traceable in a single audit trail — not reconstructed manually from four separate systems before an inspection.
If your quality system isn't doing that, the five signs above aren't isolated problems. They're symptoms of the same underlying gap: a quality infrastructure that was built for a simpler organization and hasn't scaled with the complexity, the headcount, or the regulatory expectations your team is now working under.
How to Diagnose Where Your Quality System Is Failing
Before evaluating platforms or planning a migration, the right first step is an honest gap analysis. Specifically across five domains: document control and version management, CAPA effectiveness, training compliance linkage, deviation traceability, and audit readiness.
The questions that usually surface the most significant gaps:
How long would it take to produce complete CAPA documentation for the last two years, including root cause, corrective action, and effectiveness verification, if an auditor asked for it today?
Can you generate a current training compliance report — showing who has acknowledged which SOP version — in under ten minutes without touching a spreadsheet?
If a deviation is discovered today, how long does it take to pull every related record: the SOP in effect at the time, the training records of the staff involved, the equipment calibration status, and the CAPA history for similar events?
When was the last repeat audit finding? What was the root cause — and was it actually resolved, or documented as resolved?
If those questions are uncomfortable to answer, that discomfort is information. It tells you exactly where your quality system's ceiling is — and where your compliance risk is concentrated.
The Bottom Line
A quality management system that works is invisible to auditors because everything is where it should be. It's visible to your team because it makes the right action easier than the wrong one. And it's measurable in outcomes: fewer repeat findings, faster audit cycles, less administrative overhead per quality event, and a quality team that spends its time on improvement work instead of system management.
If any of the five signs above describes your current environment, the question isn't whether your quality system is holding your organization back. It's how much further back it will fall before the gap gets addressed.
Related: