
Quality management built for the rigor of pharmaceutical and biotech manufacturing
Pharmaceutical and biotech manufacturers face a quality challenge that general-purpose software wasn't designed for. FDA, EMA, PMDA, and ANVISA simultaneously. Batch records that need to connect to deviations that connect to CAPAs that connect to change control. Annual product reviews that require data from a dozen sources. Inspections that treat every documentation gap as a finding. Kintavo gives pharmaceutical and biotech quality teams a single GMP-compliant platform to manage all of it — built for the agency scrutiny, the batch complexity, and the regulatory expectations that come with making products people put in their bodies.
One platform for every quality domain a GMP manufacturer manages.
GMP-compliant quality for branded, generic, specialty pharma, and biologics — built for FDA, EMA, and PMDA scrutiny.
Document Control
Master batch records, SOPs, specifications, and validation protocols with version control and electronic signatures.
Learn moreDeviation Management
Structured deviation capture with severity classification, investigation workflow, and CAPA linkage.
Learn moreCAPA Management
Root-cause analysis, corrective action tracking, and effectiveness verification — with reopen triggers when actions don't hold.
Learn moreChange Control
Impact assessment, multi-tier approval routing, validation gating, and post-implementation effectiveness checks.
Learn moreTraining Management
Version-linked GMP role-based training that retires automatically when SOPs revise.
Learn moreAudit Management
Internal audits, supplier audits, and inspection readiness across FDA, EMA, PMDA, and ANVISA expectations.
Learn moreQC Analysis
OOS investigations, trending against control limits, and APR/PQR data aggregation.
Learn moreSupplier Quality
API supplier qualification, excipient vendor monitoring, and CMO oversight workflows.
Learn more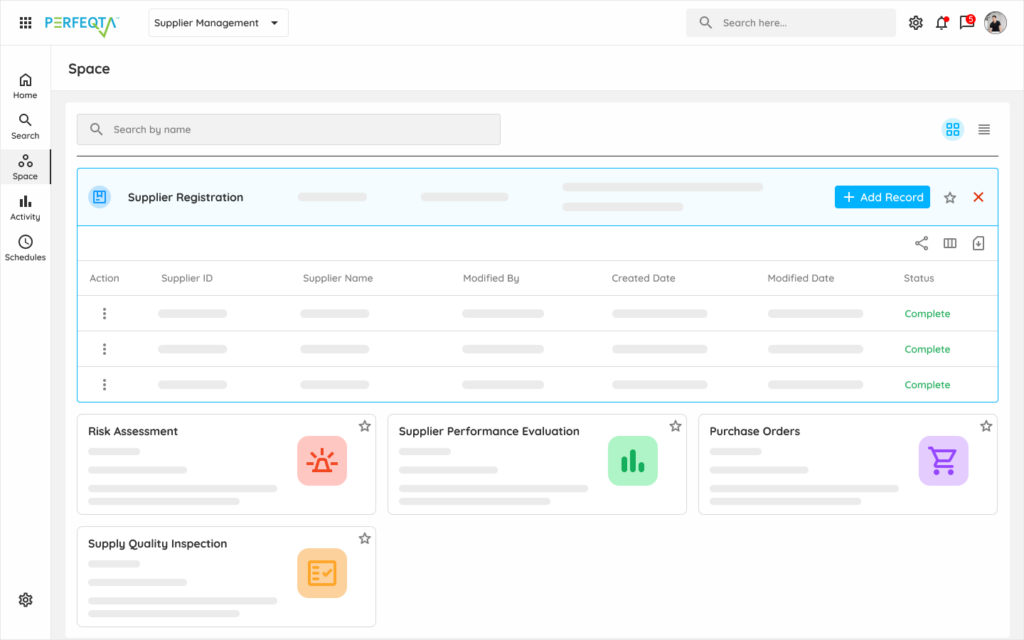
Compliance coverage for pharmaceutical and biotech manufacturing.
Same platform, ready for whichever agency walked in this morning.
FDA cGMP for finished pharmaceuticals and APIs.
EudraLex Volume 4 — European GMP guidelines including Annex 1 (sterile) and Annex 15 (qualification).
Pharmaceutical quality system framework across product lifecycle.
Quality risk management principles applied to deviations and changes.
GMP for active pharmaceutical ingredients.
FDA electronic records and signatures for batch records and quality data.
Security and availability controls for batch and quality data.
One system, four regulators.
FDA, EMA, PMDA, ANVISA — same record, different reports.
Form 483 ready
Annex 1 compliant
J-GMP aligned
BRA-GMP ready
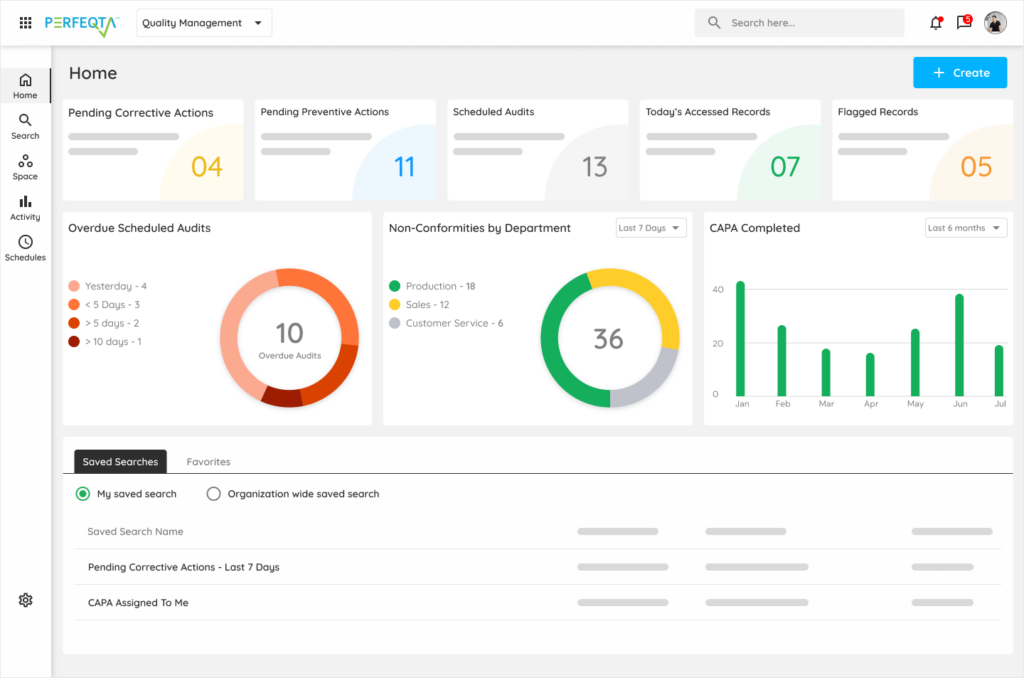
When your pharma operation runs on Kintavo.
Deviations, batch records, and CAPAs become one connected system — not three disconnected ones.
Deviations route to investigation in minutes, not days.
Batch-record exceptions tie to the CAPAs that fixed them.
Annual product reviews compile themselves from the data that already exists.
Inspections turn from fire drills into walkthroughs.
Your QA team stops writing reports about the work and starts doing the work the reports describe. The data is already there; Kintavo just gives it back.
Book your demoFAQ Questions & Answers
Q: Is Kintavo designed for pharmaceutical and biotech GMP environments? Kintavo is configured for the specific quality management requirements of GMP-regulated pharmaceutical and biotech manufacturers — 21 CFR Parts 210 and 211, EU GMP EudraLex Volume 4, ICH Q7/Q9/Q10, and 21 CFR Part 11. Document Control manages master batch records, SOPs, and validation protocols under version control with electronic signatures. Deviation Management captures batch and process nonconformances with severity classification and enforced investigation workflows. CAPA Management enforces structured root-cause analysis and effectiveness verification. The platform is purpose-built for the quality expectations that come with drug manufacturing, not adapted from a general-purpose system.
Q: How does Kintavo support FDA 21 CFR 211 compliance? Every quality system element required by 21 CFR 211 — written procedures, deviation investigations, CAPA, change control, laboratory controls, annual product reviews, equipment qualification, personnel training — is managed in Kintavo with the documentation, audit trail, and electronic signature infrastructure that satisfies both the underlying GMP requirements and the 21 CFR Part 11 electronic records requirements. When an FDA investigator submits a records request or opens a Form 483 observation, the complete evidence package is retrievable from Kintavo in minutes rather than days of manual compilation.
Q: How does Kintavo support EU GMP compliance including Annex 1? Kintavo's quality system infrastructure aligns with the requirements of EudraLex Volume 4 including Annex 1 (Manufacture of Sterile Medicinal Products) and Annex 15 (Qualification and Validation). Document Control manages validation protocols and summary reports under version control. Change Control gates process and equipment changes with impact assessment and validation hold logic. Deviation Management captures contamination control and environmental monitoring nonconformances with enforced investigation workflows. The system's audit trail, access control, and data integrity architecture meet EU Annex 11 requirements for computerized systems in GMP environments.
Q: How does change control work for GMP manufacturing processes? Kintavo's Change Control module enforces a formal change request process — including change description, scope definition, impact assessment, regulatory change evaluation, multi-tier approval routing, validation activity gating, and post-implementation effectiveness verification. A process change cannot be released to production until all required approvals and validation activities are documented and complete. Changes link automatically to the SOPs and batch records they affect, the training assignments they trigger, and the CAPAs or deviations that initiated them. The complete change history for any process or system is retrievable for regulatory submission or inspection review.
Q: How does Kintavo handle OOS investigations for pharmaceutical QC? Out-of-specification results in Kintavo trigger a structured OOS investigation workflow — Phase I laboratory investigation, Phase II full investigation with root cause, and CAPA initiation — with required fields and documented decisions at each stage. The investigation record links to the batch affected, the instrument that generated the result, the analyst who performed the test, and the specification document in effect at the time. Trending across OOS results by product, method, analyst, and instrument surfaces systemic issues that individual investigations miss. APR/PQR data for each product line aggregates automatically from historical QC and deviation records.
Q: How does Kintavo support annual product reviews and product quality reviews? Kintavo aggregates the data required for APR and PQR compilation — batch yield summaries, OOS and OOT results, deviation counts by category, CAPA closure rates, change control activity, customer complaints, stability data trending, and supplier nonconformance history — from the records that already exist in the system. What used to require weeks of manual data gathering from disconnected systems is compiled in a structured report from the source records. The APR/PQR document links to the underlying records for auditor verification.
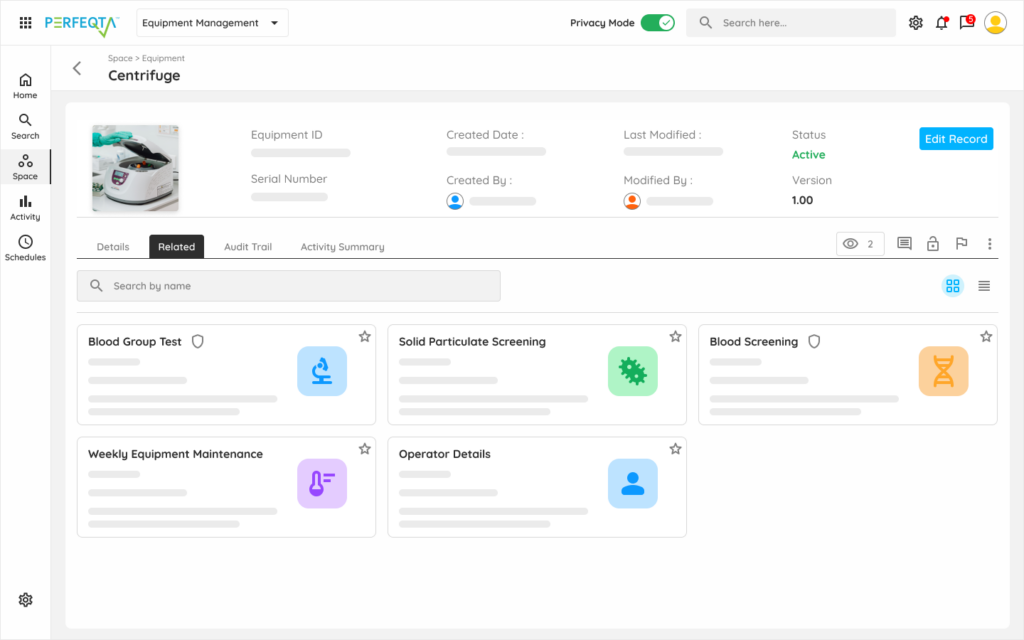
Q: How does Kintavo handle supplier qualification for pharmaceutical supply chains? Kintavo's Supplier Quality module manages API supplier qualification, excipient vendor monitoring, contract manufacturing organization oversight, and contract testing laboratory qualification workflows. Each supplier record maintains qualification status, audit history, deviation and CAPA linkages, certificate of analysis tracking, and re-qualification schedules. When a supplier generates a deviation or nonconformance, the impact on affected batches and the CAPA response are both tracked in the same record. The approved supplier list is current in real time — not in a spreadsheet updated manually when someone remembers.
Q: Is Kintavo validated as a GMP computerized system? Kintavo is designed and maintained as a GxP-regulated computerized system meeting the requirements of 21 CFR Part 11, EU Annex 11, and GAMP 5. Kintavo provides IQ/OQ documentation to support customer validation activities. The platform's change control process, audit trail completeness, access control architecture, and data integrity controls are built to satisfy the computerized system expectations of FDA, EMA, and PMDA inspectors. Customer organizations are responsible for their own OQ/PQ validation activities, which Kintavo's documentation package is designed to support efficiently.
Q: Can Kintavo support multi-site pharmaceutical manufacturing networks? Yes. Pharmaceutical manufacturers operating across multiple drug product manufacturing sites, API manufacturing facilities, packaging operations, and distribution centers can be managed under one Kintavo tenant with site-specific permissions, workflow configurations, and reporting. Each site operates with its own quality workflows while sharing the same compliance infrastructure. Corporate quality leadership sees consolidated metrics, trending data, and inspection readiness scores across all sites without requiring separate systems or manual data aggregation from each facility.
Q: How does Kintavo support inspection readiness for multi-agency manufacturers? Kintavo's Audit Intelligence™ continuously monitors your quality records against configurable regulatory frameworks — FDA 21 CFR 211, EU GMP, ICH Q10, and others — scoring your readiness in real time and surfacing likely findings before an inspector does. Pre-inspection reports pull current evidence across every module: SOP currency, training completion rates, open deviation and CAPA counts, change control backlog, equipment qualification status, and supplier qualification currency. For manufacturers subject to multiple regulatory agencies simultaneously, the same record set satisfies all frameworks — different inspection prep reports from the same underlying data.
Q: What does implementation look like for a pharmaceutical or biotech manufacturer? Your implementation lead works with your quality team to configure deviation categories, CAPA workflows, change control gating logic, document approval routing, training curricula, QC programs, equipment records, supplier qualification workflows, and regulatory framework mappings — typically within the first two to three weeks of onboarding. Kintavo provides IQ/OQ documentation to support your GxP computerized system validation. Historical SOPs, open quality records, and batch data can be migrated from spreadsheets or prior systems. Most pharmaceutical manufacturers are processing live quality events in Kintavo within 30 days of kickoff.
GMP scrutiny is unforgiving.
FDA, EMA, PMDA, ANVISA — each agency reads the same regulation differently. Each inspection finds what the last one missed. Branded, generic, specialty, biologic — every modality carries its own quality expectations, its own validation rigor, its own post-market signals.
Managing that on systems built for general manufacturing is how 483s happen. Kintavo is purpose-built for the difference.