
Quality management built for the full complexity of medical device development and manufacturing
Medical device quality programs don't fit neatly into a single regulatory lane. Design controls under 21 CFR Part 820. Risk management under ISO 14971. Post-market surveillance feeding both. EU MDR adding a new layer on top. Software adding IEC 62304. UDI adding a database the FDA reads automatically. The teams that try to manage all of it across PLM, Jira, and Excel discover at audit time how much fell between the tools. Kintavo gives medical device quality teams a single system where design history files, risk management files, CAPAs, change controls, and post-market surveillance live together — connected in one record, not reconciled from four.
One platform for every quality domain a device manufacturer manages.
ISO 13485 and 21 CFR Part 820 out of the box — with design controls, DHF management, and post-market surveillance.
Document Control
Design history files, device master records, technical files, and SOPs with version-linked training.
Learn moreChange Control
Engineering change orders with impact assessment, design-output traceability, and validation gating.
Learn moreRisk Management
ISO 14971-aligned risk management file, hazard identification, and risk-control measure tracking.
Learn moreCAPA Management
Corrective and preventive action workflows tied to complaints, MDRs, audit findings, and design-control deviations.
Learn moreTraining Management
Role-based training for design, manufacturing, and post-market staff with competency verification.
Learn moreSupplier Quality
Component supplier qualification, sterilization vendor oversight, and contract manufacturer agreements.
Learn moreAudit Management
ISO 13485 surveillance, Notified Body audits, FDA inspections, and internal quality system audits.
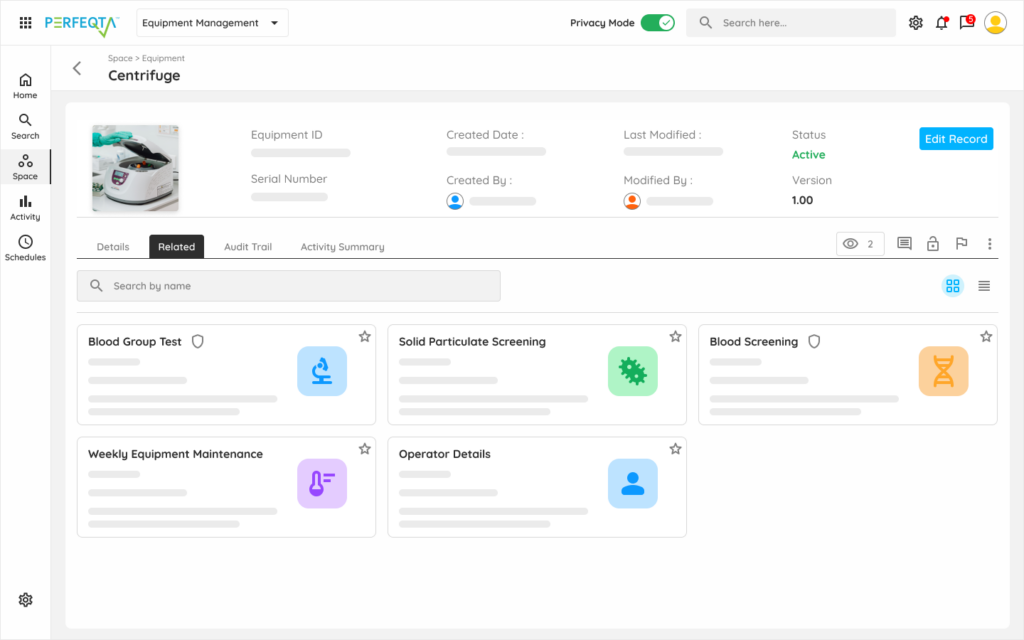
Learn moreEquipment Management
Production equipment qualification, calibration, and preventive maintenance for manufacturing assets.
Learn more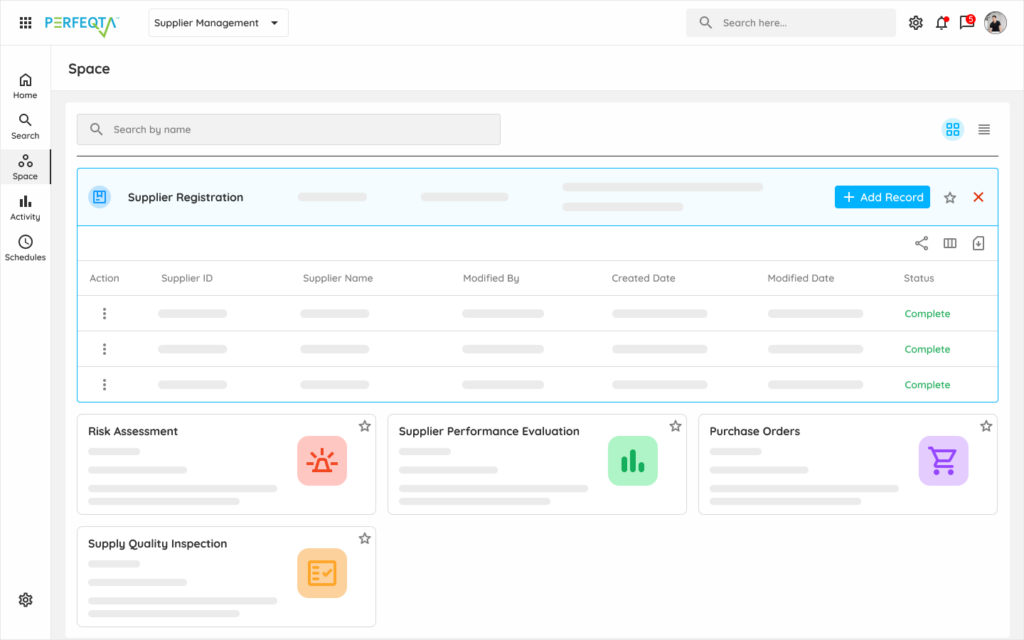
Compliance coverage for medical device quality management.
Design controls, risk management, and post-market surveillance — under one quality system.
Quality management systems for medical device organizations.
FDA Quality System Regulation for medical devices.
Medical Device Regulation (EU) 2017/745.
Risk management for medical devices.
Software lifecycle processes for medical device software.
FDA electronic records and signatures.
Unique Device Identification labeling and database requirements.
DHF that stays alive past submission.
Design inputs, outputs, V&V, risk file — linked to the CAPAs and changes that touch them.
When your device program runs on Kintavo.
Design controls, risk management, and post-market surveillance become one connected file — instead of three disconnected ones.
DHF stays alive across the product lifecycle, not frozen at submission.
Risk-control measures link to the design outputs they verify.
Post-market surveillance feeds the next risk assessment automatically.
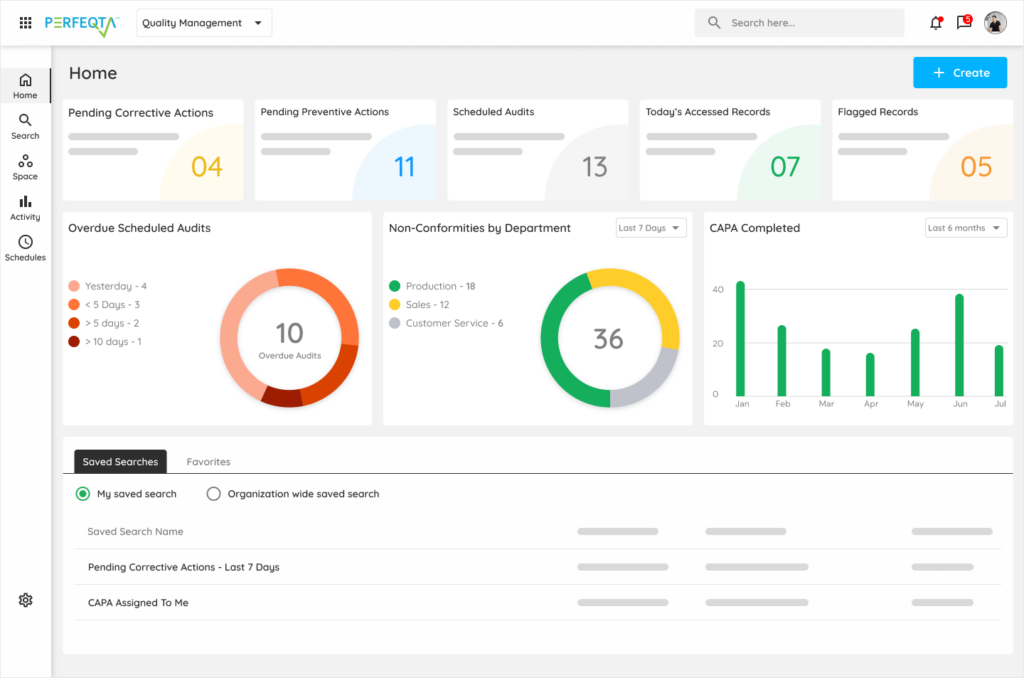
ISO 13485 audits surface what's working, not what's missing.
Your design and quality engineers stop reconciling PLM, Jira, and Excel and start reading one record — the one that actually represents the device today.
Book your demoFAQ Questions & Answers
Q: Is Kintavo designed for medical device quality management systems? Kintavo is configured for the specific requirements of medical device quality management — ISO 13485, 21 CFR Part 820, EU MDR, ISO 14971, and 21 CFR Part 11. Document Control manages design history files, device master records, and technical files under version control with electronic signatures. Change Control manages engineering change orders with design-output traceability and validation gating. Risk Management supports ISO 14971-aligned hazard identification and risk-control measure tracking. The platform is designed to keep design controls, risk management, and post-market surveillance connected in one system rather than fragmented across PLM, Jira, and Excel.
Q: How does Kintavo support ISO 13485 certification? Kintavo's quality system infrastructure covers every clause of ISO 13485 — document control, records management, management review data, product realization, measurement and monitoring, and corrective and preventive action. Audit Intelligence™ continuously monitors your quality records against ISO 13485 requirements, surfacing gaps before a surveillance audit. Pre-audit reports identify open items mapped to specific ISO 13485 clauses. Internal audit workflows and Notified Body audit preparation both run through the same system. The complete quality system evidence for ISO 13485 certification and annual surveillance is maintained in Kintavo and retrievable in minutes.
Q: How does Kintavo support 21 CFR Part 820 compliance? Every element of the FDA Quality System Regulation — design controls, document control, purchasing controls, production and process controls, corrective and preventive action, complaint handling, and records — is managed in Kintavo with the audit trail, electronic signature infrastructure, and enforcement logic that satisfies 21 CFR Part 820 and 21 CFR Part 11. When an FDA investigator submits a records request during an inspection, the complete evidence package is retrievable from Kintavo without manual compilation from disparate systems.
Q: How does Kintavo support EU MDR compliance? EU MDR 2017/745 introduced Post-Market Surveillance plans, Periodic Safety Update Reports, and enhanced technical documentation requirements on top of the existing ISO 13485 quality system framework. Kintavo manages PMS data capture — complaints, vigilance reports, post-market clinical follow-up — with linkage to the risk management file and design documentation. Change controls for design modifications include EU MDR impact assessment workflows. The technical documentation for each device family is maintained under version control and accessible for Notified Body review.
Q: How does risk management work for medical devices in Kintavo? Kintavo's Risk Management module supports ISO 14971-aligned risk management — hazard identification, risk estimation, risk evaluation, risk control, and residual risk assessment — for medical device design and manufacturing processes. Risk records link to the design outputs that implement risk controls, the test records that verify their effectiveness, and the post-market surveillance data that monitors residual risk in the field. When a complaint trend or CAPA surfaces new information relevant to a known hazard, the risk record updates automatically. The complete risk management file is traceable from initial hazard identification through post-market monitoring.
Q: How does design history file management work in Kintavo? The DHF in Kintavo is not a static document collection — it's a live record that stays current as the device evolves. Design inputs, design outputs, verification and validation records, design reviews, and design transfer documents are maintained under version control with electronic signatures. Engineering change orders link to the design documents they affect, so the DHF reflects what the device actually is today, not what it was at 510(k) submission. When a CAPA or complaint identifies a design issue, the linkage from post-market event back to the relevant design output is traceable in the same system.
Q: How does Kintavo handle post-market surveillance and complaint management? PMS data in Kintavo — complaints, MDRs, vigilance reports, post-market clinical follow-up, and field safety corrective action records — links directly to the device's risk management file and CAPA system. Complaint trends trigger structured investigations with required fields and documented disposition decisions. When a complaint warrants an MDR or vigilance report, Kintavo routes the record through a regulatory reporting review workflow before closure. The post-market surveillance data feeds the next periodic review of the risk management file, creating a continuous loop between field data and design documentation.
Q: How does change control work for medical device engineering changes? Kintavo's Change Control module manages engineering change orders with impact assessment across the technical documentation, risk management file, and validation status of affected processes and equipment. A design change cannot be released to manufacturing until all required impact assessments, design verification activities, and approval signatures are documented and complete. Changes link to the DHF records they affect, the training assignments they trigger, and the risk management records that need to be updated. The complete change history for any design element is retrievable for FDA inspection or Notified Body review.
Q: Can Kintavo support IEC 62304 software lifecycle requirements? Yes. Kintavo's Document Control and Change Control modules manage the software lifecycle documentation required by IEC 62304 — software development plans, architecture documentation, unit implementation and verification records, software integration testing, and software release documentation — under version control with electronic signatures. Software changes follow the same change control workflow as hardware changes, with IEC 62304 safety classification driving the required review and verification activities. Software problem reports link to the CAPA system for systematic issue tracking and resolution.
Q: How does supplier qualification work for medical device supply chains? Kintavo's Supplier Quality module manages component supplier qualification, sterilization vendor oversight, contract manufacturer agreements, and critical service provider qualification — all within the same system as your internal quality records. Each supplier record maintains qualification status, audit history, nonconformance and CAPA linkages, and re-qualification schedules. Supplier audits use the same checklist and finding capture infrastructure as internal audits. When a supplier nonconformance affects a device lot, the impact on in-market devices and the corrective action response are both tracked in one traceable record.
Q: What does implementation look like for a medical device manufacturer? Your implementation lead works with your quality team to configure document control workflows, DHF structure, risk management templates, CAPA processes, change control gating, training curricula, supplier qualification workflows, and regulatory framework mappings — typically within the first two to three weeks of onboarding. Existing design history files, open CAPAs, risk management files, and supplier records can be migrated from spreadsheets or prior systems. Kintavo provides IQ/OQ documentation to support computerized system validation under ISO 13485 and 21 CFR Part 820. Most device manufacturers are processing live quality events in Kintavo within 30 days of kickoff.
Device quality requires three systems pretending to be one.
Design controls under 820.30. Risk management under ISO 14971. Post-market surveillance feeding both. EU MDR added a new layer on top. Software added IEC 62304. UDI added a database the FDA reads automatically.
The teams that try to manage this in PLM, Jira, and Excel discover at audit time how much fell between the tools. Kintavo is the single record the audit was always looking for.